P09: Development of software for better assessment of non-coding variants on gene expression
Principal investigators: Prof. Dr. Dominik Seelow and Prof. Dr. Markus Schuelke
 While much progress has been achieved in predicting the deleteriousness of coding variants, the assessment of non-coding variants still leaves much to be desired. With only about a third of current Exome Sequencing projects yielding a causal variant, it has become clear that the non-coding space also plays a role in genetic disease. For example, it is self-evident that the homozygous disruption of a gene promoter can lead to the complete loss of protein expression and has hence the same consequence as a homozygous nonsense variant.
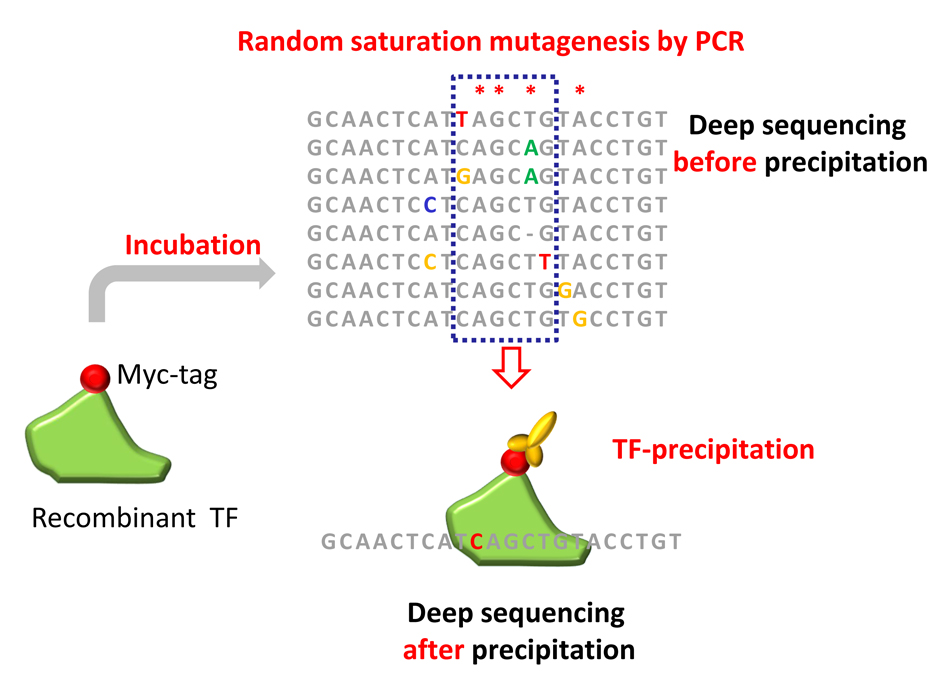
In the first funding period, we have focused on predicting the quantitative effects of variants on transcription factor binding strength and demonstrated that neural networks can identify hitherto unknown binding motifs and give reliable estimates of variant effects on transcription factor binding affinity. This work was a collaboration with the former project P03 (Heinemann/Schmidt-Ott).
In the upcoming funding period, we plan to develop software that combines deep learning-based models for transcription factor binding sites with information about regulatory regions in the genome, hence allowing us to predict whether DNA variants are located within promoters or enhancers of disease-relevant genes and to what extent they might affect transcription factor binding.
We will also create a command-line tool to be integrated into genome sequencing pipelines, as well as a web-based application for researchers to test potentially regulatory variants for potential effects on functional candidate genes.
We will integrate these functionalities into our mutation detection software, MutationDistiller, which ranks DNA variants based on their deleteriousness to the gene and the role of the affected gene in the patient's phenotype. We hope that this endeavor will help to achieve the main goal of this Research Unit: To shorten or even eliminate the diagnostic odyssey of patients with rare genetic disorders.
While much progress has been achieved in predicting the deleteriousness of coding variants, the assessment of non-coding variants still leaves much to be desired. With only about a third of current Exome Sequencing projects yielding a causal variant, it has become clear that the non-coding space also plays a role in genetic disease. For example, it is self-evident that the homozygous disruption of a gene promoter can lead to the complete loss of protein expression and has hence the same consequence as a homozygous nonsense variant.
In the first funding period, we have focused on predicting the quantitative effects of variants on transcription factor binding strength and demonstrated that neural networks can identify hitherto unknown binding motifs and give reliable estimates of variant effects on transcription factor binding affinity. This work was a collaboration with the former project P03 (Heinemann/Schmidt-Ott).
In the upcoming funding period, we plan to develop software that combines deep learning-based models for transcription factor binding sites with information about regulatory regions in the genome, hence allowing us to predict whether DNA variants are located within promoters or enhancers of disease-relevant genes and to what extent they might affect transcription factor binding.
We will also create a command-line tool to be integrated into genome sequencing pipelines, as well as a web-based application for researchers to test potentially regulatory variants for potential effects on functional candidate genes.
We will integrate these functionalities into our mutation detection software, MutationDistiller, which ranks DNA variants based on their deleteriousness to the gene and the role of the affected gene in the patient's phenotype. We hope that this endeavor will help to achieve the main goal of this Research Unit: To shorten or even eliminate the diagnostic odyssey of patients with rare genetic disorders.